TNF is a pleiotropic cytokine serving important functions in pathophysiology, as it plays regulatory role in the growth, differentiation and death of immune and non-immune cells. Its functions are mediated through binding to two transmembrane receptors, TNFR1 and TNFR2, that deliver signals to activate a variety of responses ranging from proliferation to apoptosis and necrosis.
Dysregulation of TNF has been implicated in immune mediated inflammatory diseases including rheumatoid arthritis, inflammatory bowel disease, psoriasis, scleroderma, systemic lupus erythematosus, atherosclerosis as well as autoimmune pathologies, cancer etc.
Since the approval of the first TNF inhibitors, back in 1998, TNF blockade has been used as a successful approach in treating a broad range of pathologies, having nonetheless the expected compromises stemming from the severe side effects that result from the blockade of the whole range of the physiological functions of TNF, with the increased susceptibility to infections being one of the majordrawbacks of this therapeutic approach.
The pioneering work of two Greek scientists, Kassiotis and Kollias, published in 2001 in JEM (193:427) and JCI (107:1507), shed light for the first time on the importance ofuncoupling the TNFR1- and TNFR2-TNF signaling pathways. This work proposed that specific blockade of TNFR1 signaling could offer advantageous novel therapeutic approaches as they would suppress the TNF pro-inflammatory activity, leaving unaffected its TNFR2-dependent immunosuppressive activity. In the following years, indeed TNFR1 emerged as an attractive target towards ameliorating the detrimental effects of the TNF signalling pathway and various TNFR1 inhibitors are currently under development or in clinical trials.
A unique tool for testing anti-hTNFR1 therapeutics is the human TNFR1 knock in (hTNFR1KI) mouse model, that expresses human TNFR1 in the absence of its mouse counterpart. The hTNFR1KI mouse combined, as shown in the table below, with induced or spontaneous disease models offers a fully equipped armory for the evaluation of human therapeutics targeting TNFR1.
Preclinical Platforms for the evaluation therapeutics targeting human TNFR1
| Model | Duration | Read outs |
| Experimental Autoimmune Encephalomyelitis
MOG-induced EAE in hTNFR1KI mice |
25-30 days | -Weight
-Clinical Scoring -Histology |
| Spontaneous Arthritis
Tg197hTNFR1KI TNFΔARE/+ hTNFR1KI |
12 weeks
4-8 weeks |
-Weight
-Clinical Scoring -Histology |
| Induced Arthritis
Collagen Antibody Induced Arthritis (CAIA) in hTNFR1KI mice |
2 weeks | -Weight
-Clinical Scoring -Histology -Serum Cytokine levels |
| Acute Inflammation
TNF/GaIN induced inflammation in hTNFR1KI mice |
24 hours | -Serum cytokine levels-Survival |
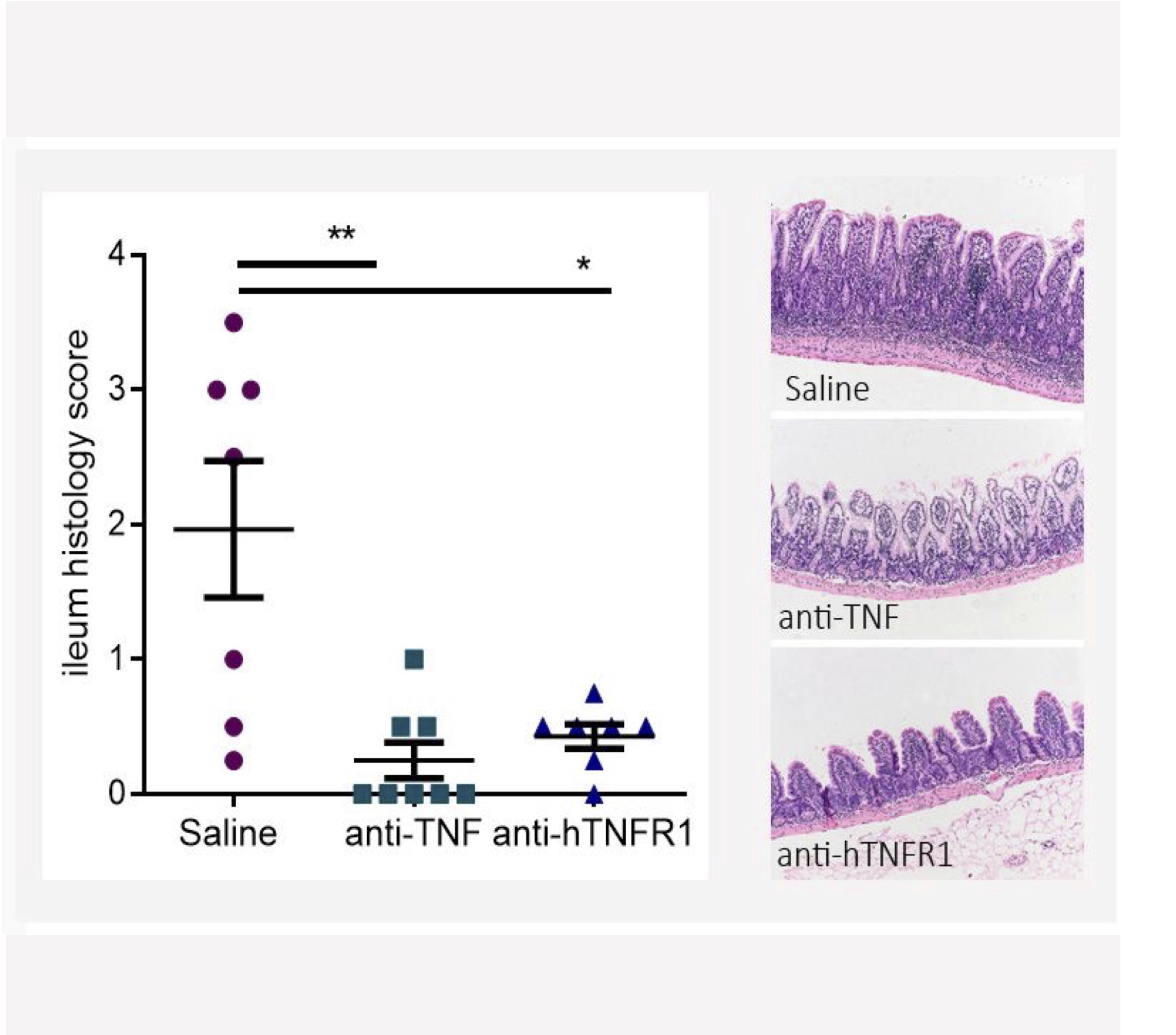
One of the models described above that has a particular interest due to its complexity and similarity to the human condition is the TNFΔAREhTNFR1KI mouse model that due to the dysregulated expression of TNF develops spontaneously intestinal inflammation, arthritis and cardiovascular disease, perfectly recapitulating many of the features that constitute the complexity of the human disease. TNFΔARE was the first mouse model that unequivocally established the causal role of TNF in the inflammatory response of Crohn’s-like ileitis, while further studies revealed numerous promising therapeutic targets some of which were later developed into approved drugs, such as Ustekinumab, vedolizumab, and natalizumab.
The TNFΔAREhTNFR1KI mouse model, faithfully reproduces all TNFΔARE phenotypes and is an ideal tool for translational research, supporting the drug discovery and development of biologics and small molecules targeting TNF, hTNFR1 as well as other potential therapeutic targets of Crohn’s-like ileitis.